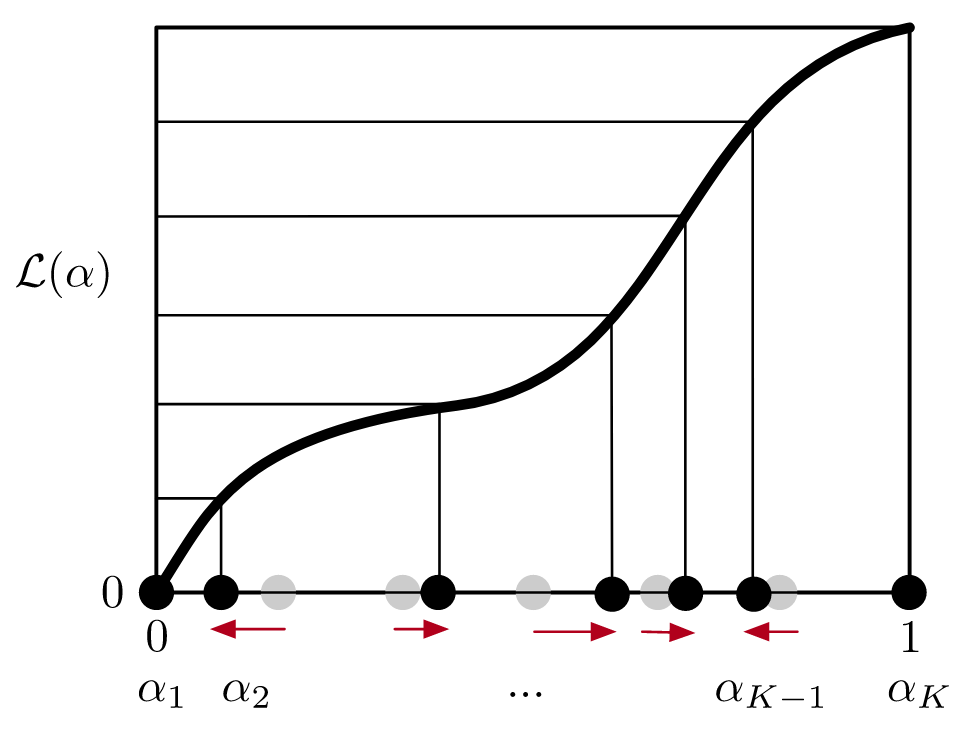
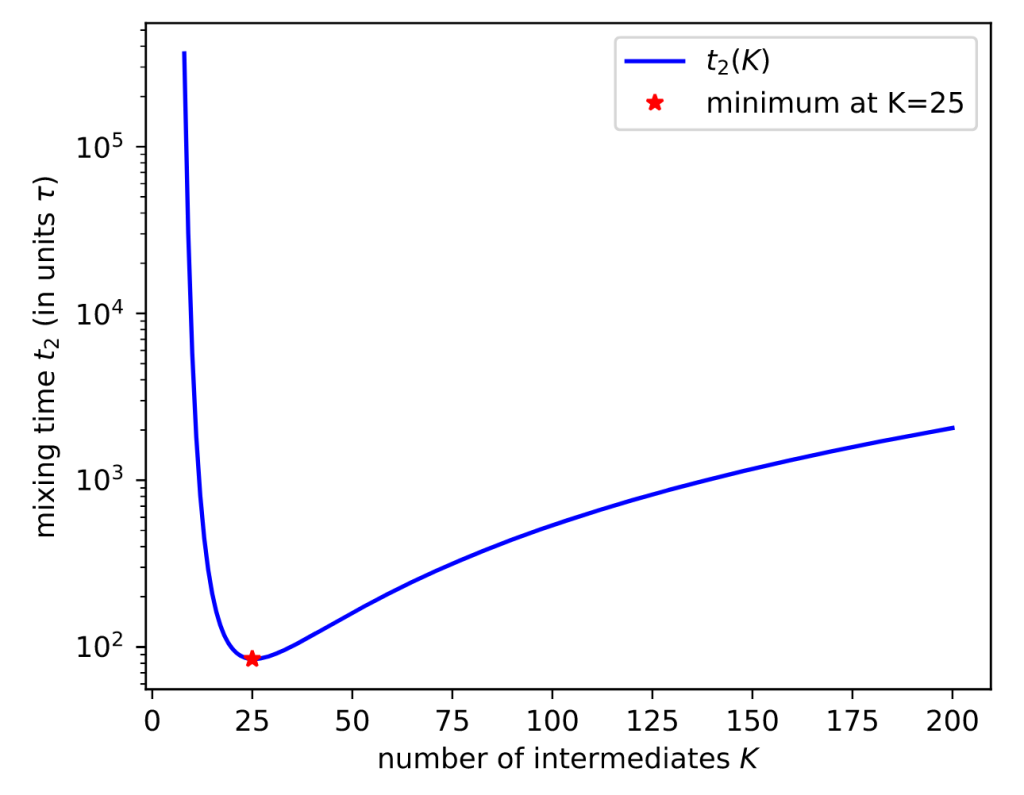
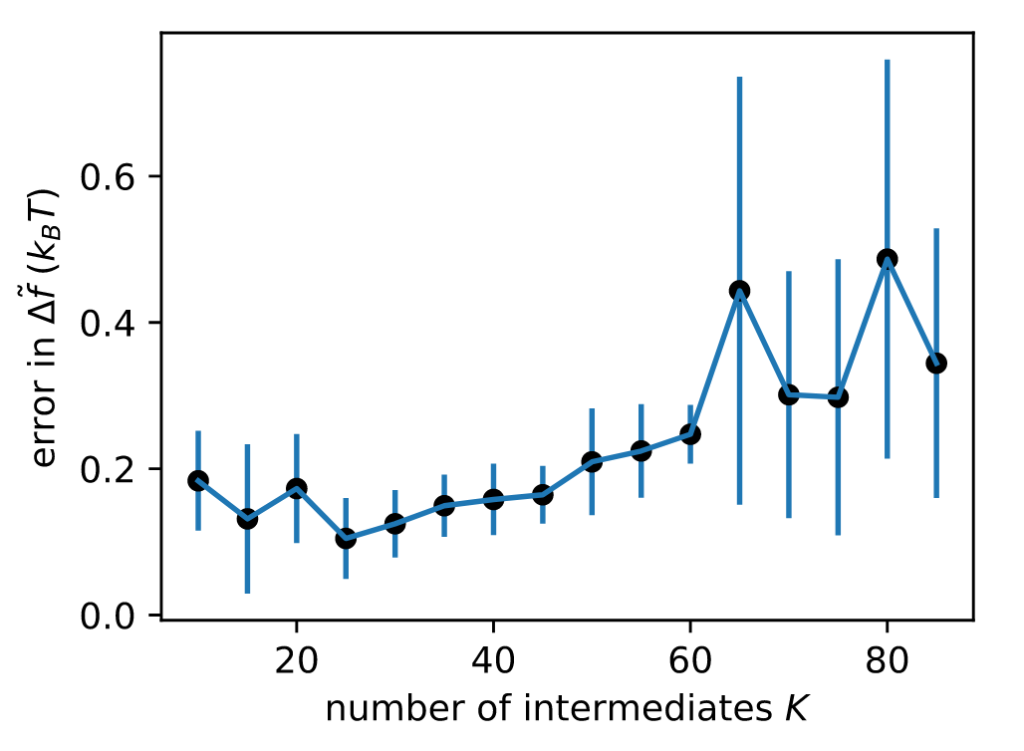
In collaboration with Jiwon Seo‘s group at GIST, we are pleased to be co-authors on a Just Accepted manuscript in Organic Letters:
Noncanonical Folding of Peptoid Oligomers: Formation of a Closed Conformation in Nonpolar Solvent. Jinyoung Oh, Min June Yang, Xingyu Chen, Juhye Shin, Bradley S. Harris, Robert M. Raddi, Suhyun Park, Marcel D. Baer, Hohjai Lee, Chin-Ju Park, Vincent A. Voelz, and Jiwon Seo. Organic Letters, ahead of print, June 22, 2026. https://doi.org/10.1021/acs.orglett.6c02040.
Peptoids (N-subsituted oligoglycines) are versatile peptide mimetics that can fold into specific three-dimensional structures.
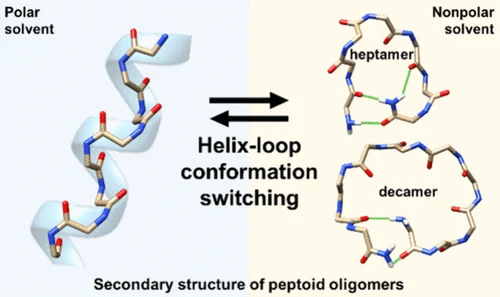
In this work, the Seo group used extensive NMR spectroscopy to study an number of N-(S)-1-phenylethylglycine (Nspe) oligomers in chloroform. Nspe homooligomers were already known to form helices in polar solvent, and an Nspe nonamer to form a “threaded loop” structure in acetonitrile. After systematically investigating a series of oligomers (Nspe)n ranging from lengths n=3 to n=12, the Seo group surprinsgly found evidence that (Nspe)7 and (Nspe)10 folded to distinct conformations in chloroform.
To solve the structure of the folded peptoids, we performed Hamitonian replica exchange molecular dynamics simulations with NMR-derived distance restraints using GROMACS, followed by conformational clustering and reweighting of simulated ensembles agasint the experimental data using the BICePs algorithm. For the force field , Marcel Baer’s group at PNNL provided the excellent STEPs-SOL, a peptoid-specific parameterization of GAFF that includes solvent effects.
The BICePs results reveal structures of (Nspe)7 and (Nspe)10 in chloroform reminiscent of the “threaded loop” structure, stabilized by end-to-end interactions that mask polar surfaces, with specific patterns of cis and trans amides along the chain. This chameleonic behavior (folding to different structures in polar vs. nonpolar media) is characteristic of macrocyclic compounds that can permeate membranes, and indeed, PAMPA measurements show marginal membrane permeability for (Nspe)7.
This work adds to the growing catalog of peptoid sequences with folded structures. A long-term hope is that continued elaboration of sequence-structure relationships for peptoids, using a combination of simulation and experiment, will lead to better (and perhaps AI-enabled) computational algorithms to design peptoids with specific molecular architectures and functions.