Raddi, Robert M., Yunhui Ge, and Vincent A. Voelz. “BICePs v2.0: Software for Ensemble Reweighting Using Bayesian Inference of Conformational Populations.” Journal of Chemical Information and Modeling 63, no. 8 (April 24, 2023): 2370–81. https://doi.org/10.1021/acs.jcim.2c01296.
Congrats to Rob Raddi on this paper and for coding this more user-friendly and extensible Python package: https://github.com/vvoelz/biceps
With five students presenting at BPS, the Voelz Lab made a strong showing at BPS 2023 in San Diego, CA, Feb 18-22! Dylan Novack gave an early-morning (yet still riveting 🙂 Platform Session talk on work simulating FOXO1 and the effects of mutations. Here are the presentations:
An Evaluation of Force Field Accuracy for the Mini-Protein Chignolin using Markov State Models Tim Marshall, Robert Raddi, Vincent A. Voelz
Model selection using Bayesian inference of conformational populations with replica averaging Robert M. Raddi, Tim Marshall, Yunhui Ge, and Vincent A. Voelz
ONCOGENIC MUTATIONS IN THE DNA-BINDING DOMAIN OF FOXO1 THAT DISRUPT FOLDING: QUANTITATIVE INSIGHTS FROM EXPERIMENTS AND MOLECULAR SIMULATION. Dylan Novack Platform Session Title: Platform: Protein Stability, Folding, and Chaperones
De novo design of beta hairpin peptides with custom installed halogen-bonding groups Thi Dung Nguyen, Vincent Voelz
Computational fragment screening of dimeric SARS-CoV-2 Main Protease Rashad Reid Jr., Vincent A. Voelz
Last day of the conference, still rocking our lanyardsSpecial guest appearance by Voelz Lab alum George Pantelopulos!
Congratulations to Ph.D student Rob Raddi, a 2022 winner of the College of Science and Technology Outstanding Teaching Assistant (TA) at Temple University! This prestigious college-level award attests to Rob’s excellence in teaching. Great job and well deserved!
Our new paper “Multi-ensemble Markov Models to estimate affinities and rates of ligand binding is now published in the Journal of Chemical Physics. Congrats to Yunhui Ge for getting this across the finish line!
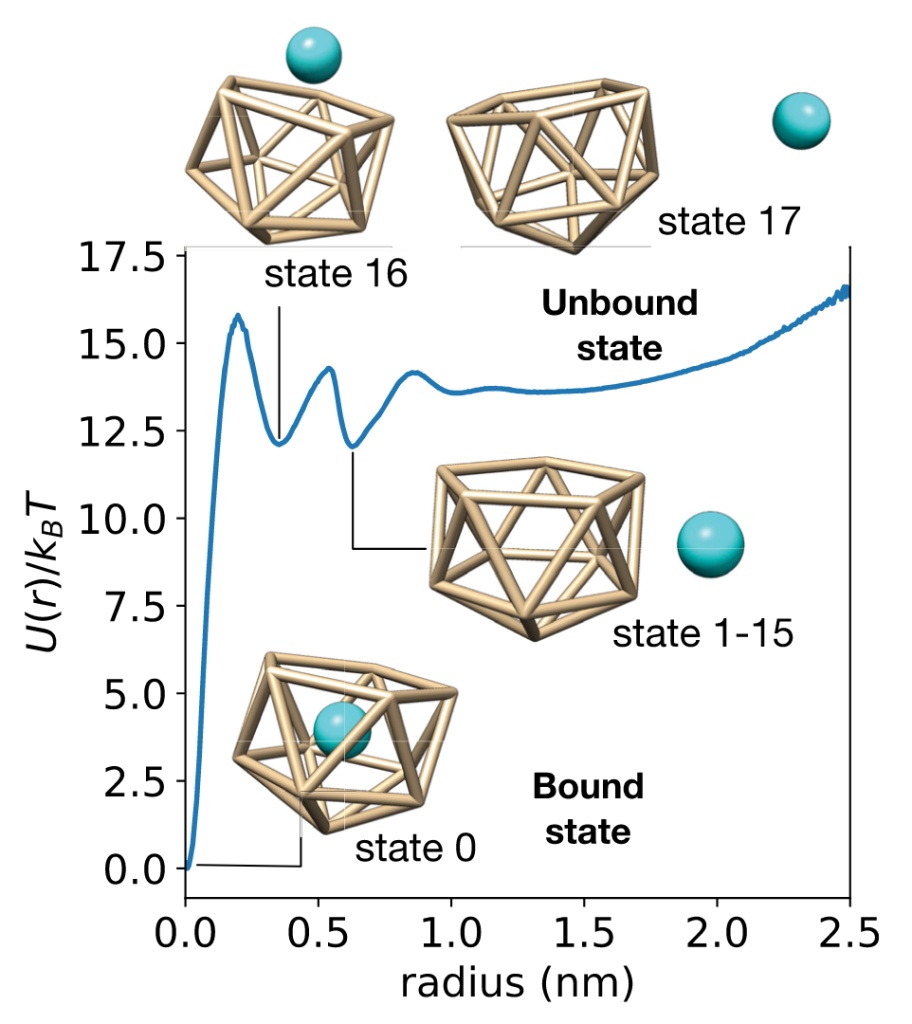
This work examines a toy receptor made from an icosahedron of 11 Lennard-Jones particles. The ligand is a LJ particle, and all is solvated in explicit water. This system has number of encounter-complex states and a slow(ish) residence time of 30.3 ns.
While conventional MSMs accurately estimate binding rates from swarms of short trajectories, they estimate affinities poorly. Why? Because MSM estimators typically enforce detailed balance, assuming the data is sampled at equilibrium.
So even with lots of parallel simulation, it still takes a long time for bound and unbound populations to equilibrate, and get a good model. (Our related work explored this problem with adaptive seeding simulations.)
Instead, what if we could quickly collect many binding/unbinding transitions in a biased ensemble, and use this information to infer populations and rates in the unbiased ensemble. This is a job for multiensemble Markov models, or MEMMs!
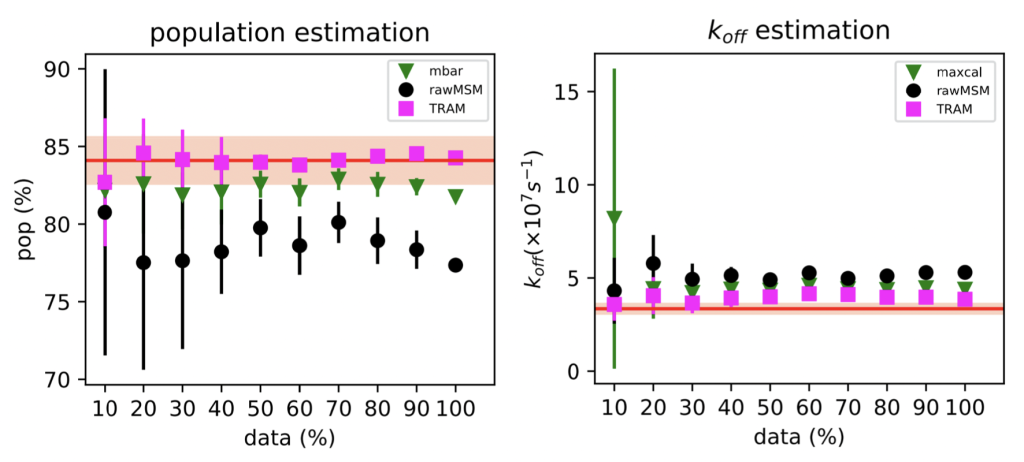
To accelerate transitions, we scaled the ligand nonbonded interactions, as is typical in FEP calculations for drug discovery. We tried two different MEMM estimators: (1) TRAM, from the Frank Noé group in Berlin, and (2) our Maximum-Caliber approach.
We find that both work pretty well (!), but TRAM works slightly better.
These results have encouraged us to explore how this method works in more realistic ligand binding systems, as a potential tool for MD-based virtual screening. Expect more from our lab on this soon 🙂
You can read more about this work here:
Estimation of binding rates and affinities from multiensemble Markov models and ligand decoupling. Ge, Yunhui, and Vincent A. Voelz. The Journal of Chemical Physics 156, no. 13 (April 7, 2022): 134115. https://doi.org/10.1063/5.0088024
Yunhui Ge, holding a model of the toy binding pocket.
On April 5, Si Zhang successfully defended her PhD thesis “Computational Approaches for Protein Folding and Ligand Binding: from Thermodynamics to Kinetics”. Congratulations Si!
Congrats to undergrad researcher and MARC scholar Rashad Reid, who presented his poster “Structural and dynamical analysis of monomeric SARS-CoV-2 Mpro active site integrity” at the Spring 2022 Meeting of the American Chemical Society in San Diego this week.
Rashad was selected for both the ACS COMP session and the SCI-MIX. Great work, Rashad!
Congrats to Voelz Lab student Si Zhang for her modeling and simulation contributions to this exciting new methodology developed by the Ross Wang Lab at Temple. These new staples use a fluorine thiol-displacement reaction for cyclization, resulting in peptides that not only are more stable their folded conformations, but are also taken up by multiple kinds of cancer cells. From large-scale folding simulations of various staple designs, Si achieved good agreement with experimental and simulated peptide helicity, suggesting that folding simulations can be a predictive tool for peptide staple design.
You can read all about it in the latest issue of Nature Communications:
Unprotected Peptide Macrocyclization and Stapling via A Fluorine-Thiol Displacement Reaction. Md Shafiqul Islam, Samuel Junod, Si Zhang, Zakey Buuh, Yifu Guan, Mi Zhao, Kishan Kaneria, Parmila Kafley, Carson Cohen, Robert Maloney, Zhigang Lyu, Vincent A. Voelz, Weidong Yang, and Rongsheng Wang. Nature Communications13, 350 (2022). https://doi.org/10.1038/s41467-022-27995-5
Students in the Voelz have been racking up a trophy case of departmental awards!
Si Zhang received a Daniel Swern Research Fellowship Award, recognizing graduate students who exhibit outstanding creativity and productivity in research.
Paige Sheridan has been awarded the Guy Allen Teaching Award for 2020-2021 Academic Year
Matthew Hurley and Si Zhang have been awarded a Spring 2022 Dissertation Completion Grant, which provides support for completing papers and their final theses.
Congratulations Paige, Si, and Matt on your excellent work to achieve these competitive and prestigious department awards!
Congrats to graduate student Si Zhang on the publication of her first-author manuscript in JCTC!
This work was a collaboration with David Hahn (Janssen Research) of Open Force Field fame, and Michael R. Shirts (University of Colorado-Boulder) who originated and implemented this expanded ensemble (EE) method in GROMACS.
We were turned on to the EE approach during our work with the COVID Moonshot, where we used CPU clients on Folding@home to screen 50k+ potential SARS-CoV-2 main protease (Mpro) inhibitors by their estimated absolute binding free energy (ABFE).
Si realized that no one had yet applied this approach to calculating relative free energies. So she tested it for a set of 24 transformations between 16 Tyk2 inhibitors using the OpenFF “Parsely” small-molecule force field, and it worked very well, achieving an accuracy within 0.66 kcal/mol (MUE) of the experimental value! Another result that portends well for using EE on distributed computing platforms like Folding@home: Performing N independent simulations gives better accuracy than a single simulation N times as long.
You can read all about the details in our hot-off-the-press paper:
Zhang, Si, David F. Hahn, Michael R. Shirts, and Vincent A. Voelz. “Expanded Ensemble Methods Can Be Used to Accurately Predict Protein-Ligand Relative Binding Free Energies.” Journal of Chemical Theory and Computation 17, no. 10 (October 12, 2021): 6536–47. https://doi.org/10.1021/acs.jctc.1c00513.