
The Liberles Research Group works in the areas of computational comparative genomics, and molecular evolution. The central theme in the research group is the detection and characterization of the lineage-specific divergence of protein-encoding genes. Much of the work in the group is done in a phylogenetic context. Ultimately, we want to ask the question, “What makes each species unique at the genomic level?” and “what are the processes driving the functional divergence of genomes?”.
Theoretical and methodological work in the group involves the construction of mechanistic models for codon and amino acid substitution and for duplicate gene retention. Ultimately, we want to ask the question, “How do proteins change functions on evolutionary timescales?”. We build mechanistic mathematical models for processes, both to ask in simulation how the processes work as a function of underlying biological parameterization and for use in inference on genome scale data.
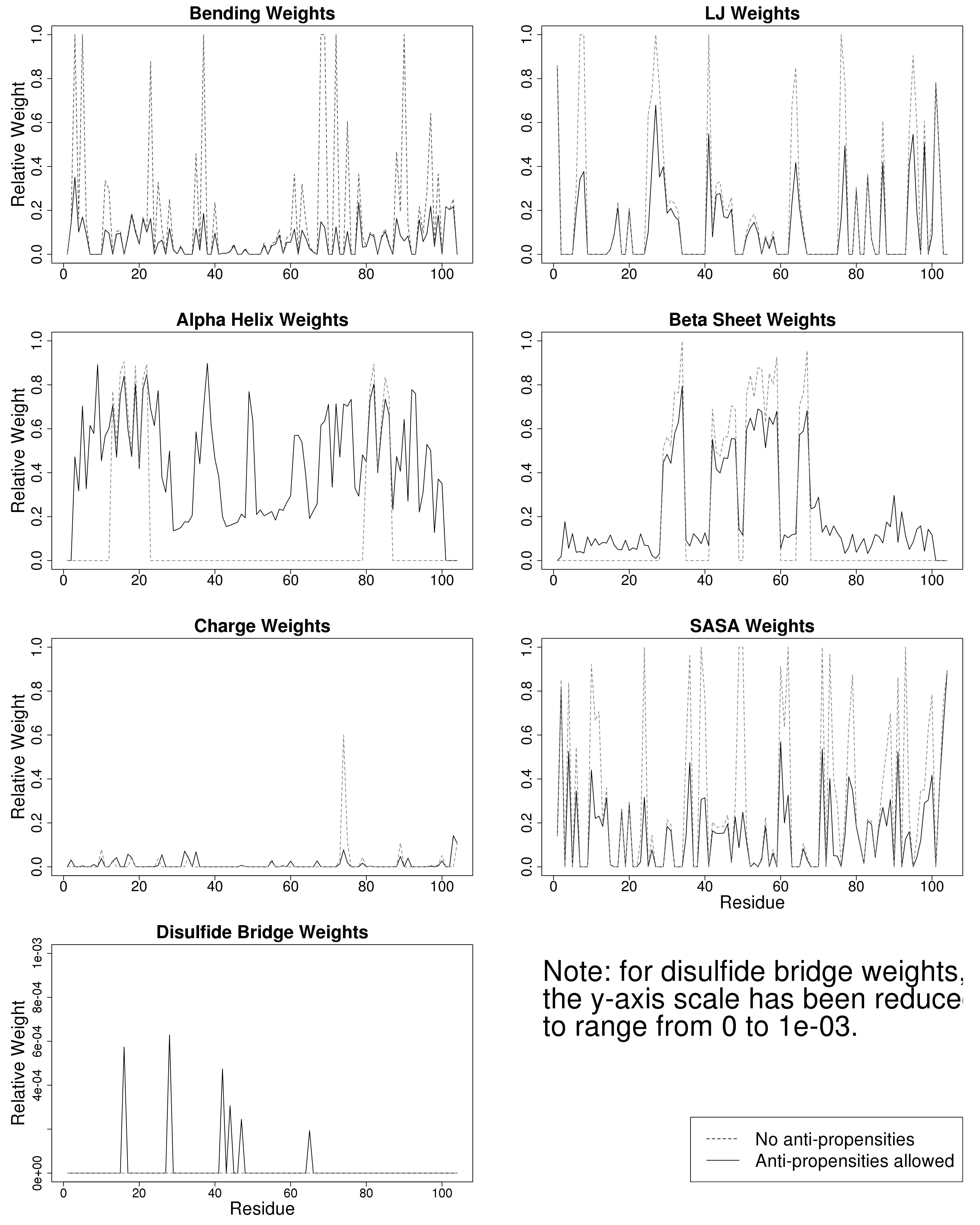
For example, the figure below on the left shows the evolutionary parameterization of a site-specific pseudoenergy function to evaluate amino acid substitution (from Proteins 86:218). The figure below on the right shows the expected time dependence of duplicate gene retention as mutations accumulate and selection acts according to combinations of different processes (neofunctionalization plus dosage constraints in A, subfunctionalization plus dosage constraints in B, from BMC EB 16:45).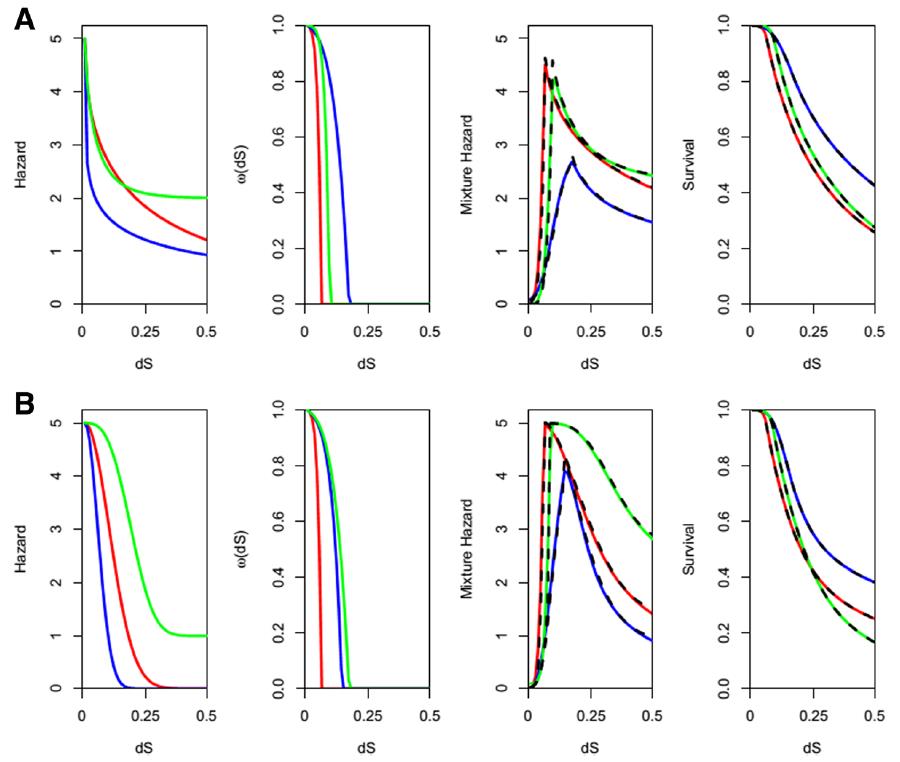
Comparative genomic work in the group has involved The Adaptive Evolution Database (TAED), where gene families and detected events of positive directional selection were mapped to species tree lineages in chordates. While the database has now been retired, we have developed some cool phylogenetic database indexing tools as well as tools for visualization of large trees in 3D hyperbolic space for this database. Recent genomic work has involved a collection of organisms, including tunicates, termites, grasses, and salmonids, among others.
Some new directions in the group include topics in evolutionary systems biology and in metagenomic data analysis to address ecological questions. Ultimately, these questions are aimed at asking, “How do proteins co-evolve when selection acts at the level of the pathway?” and “How do species interact in ecosystems and how is this linked to their gene content and genome sequences?”.
Our work in evolutionary systems biology has characterized mutation-selection-drift balance as an important process driving the evolution of individual enzymes when selection acts at the level of pathway function. This context-dependence of enzyme selective pressures has important consequences for understanding when mutations in individual proteins have phenotypic effects and when they do not. See the figure below for a view on mutation-selection-drift balance and the evolutionary half-life of flux control by individual enzymes (Biology Direct 11:31).
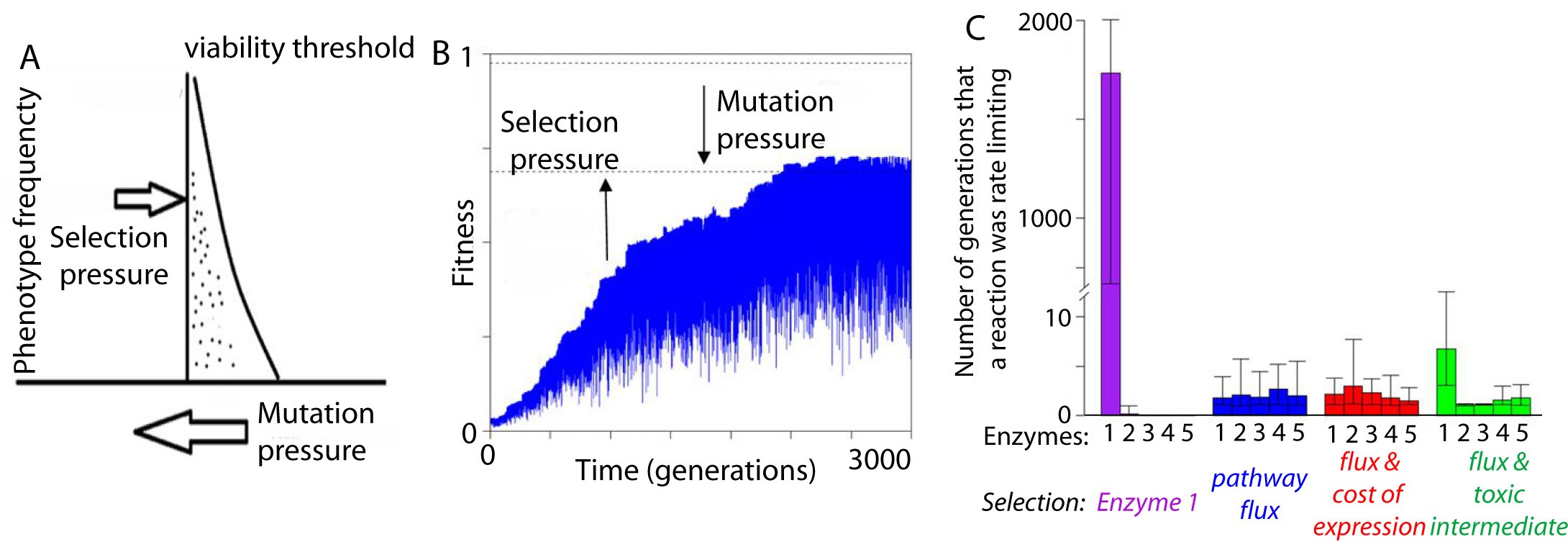
Recently, we have been working to extend our evolutionary systems biology models to detect selection on biological pathways and to be able to reconstruct ancestral states of pathway function, by applying a probabilistic process to sets of ordinary differential equations.